Maladie d’Alexander
La maladie d’Alexander est une maladie génétique rare de la famille des leucodystrophies démyélinisantes. Son incidence dans le monde n’est pas précisément connue, mais a été estimée à 1/2 700 000 au Japon. Cette maladie neurologique est diagnostiquée le plus souvent dans la petite enfance, mais des formes juvéniles et adultes existent également. Les symptômes comprennent un retard mental, l’épilepsie, une raideur musculaire, et leur progression conduit au décès du patient. Elle est caractérisée par une dégénérescence de la substance blanche et la formation de fibres de Rosenthal.
1. La mutation génétique
La maladie d’Alexander est causée par des mutations dominantes du gène GFAP qui est situé sur le chromosome 17 (en 17q21). Ce gène permet la fabrication de la protéine acide fibrillaire gliale (GFAP). Ces protéines fibrillaires s’assemblent pour former des filaments intermédiaires dans les astrocytes du système nerveux central.
En clair, c’est quoi un filament intermédiaire et leur rôle dans les astrocytes.
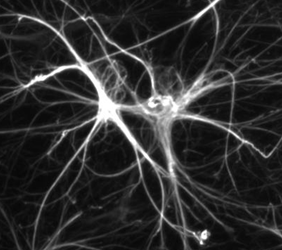
Les astrocytes sont des cellules du cerveau qui ressemblent à des astres (d’où leur nom : « astro », et « cyte » pour cellule). Elles ressemblent à des étoiles qui auraient des rayons. Ces faisceaux sont comme les bras de la cellule. Ces extensions sont nécessaires à la fonction de la cellule, qui doit échanger des informations avec beaucoup d’autres cellules de son environnement : la substance blanche.
Le cytosquelette [1], c’est le squelette de la cellule ; c’est ce qui permet à la cellule d’avoir une certaine forme.

Les filaments intermédiaires sont des constituants du cytosquelette. Ils maintiennent l’architecture de la cellule. Les filaments intermédiaires de type III sont composés de GFAP dans les astrocytes et d’autres cellules gliales. Ces nanofilaments s’assemblent spontanément et ne nécessitent ni protéine chaperonne, ni source énergétique. Comme un assemblage de lego.

Les mutations de GFAP sont dominantes, c’est-à-dire qu’une seule copie mutée du gène entraine la maladie. Dans la grande majorité des cas, la mutation est sporadique, ce qui signifie qu’elle n’est pas héritée des parents, et elle apparait pour des raisons inconnues. Déjà plus d’une centaine de variants pathogènes ont été identifiés.
Les mutations de GFAP, qui se retrouvent chez 90% des patients, sont des mutations « faux sens ». Les mutations faux sens sont des mutations ponctuelles dans lesquelles une lettre du gène est changée (un nucléotide), modifiant l’assemblage en cours de la chaine qui forme la protéine. Le résultat est une protéine modifiée, et dans la maladie d’Alexander, cette protéine gagne en activité. On parle de gain de fonction.
2. Le mécanisme simplifié conduisant à la maladie
Il est aujourd’hui clair que la maladie d’Alexander est une maladie des astrocytes. Dans les astrocytes des patients atteints de la maladie d’Alexander, la présence de GFAP mutée, et une accumulation excessive de GFAP (mutée et non mutée), sont liées à l’activation de plusieurs voies de stress cellulaire. L’hyper-activation des astrocytes pourrait être toxique et le principal contributeur de la pathogenèse. Les astrocytes sont essentiels au développement et au fonctionnement du cerveau, mais jusqu’à récemment, ces cellules neurales étaient largement ignorées. Le rôle crucial des astrocytes dans le cerveau commence maintenant à être reconnu.
Les fibres de Rosenthal
Les fibres de Rosenthal sont composées des protéines de filament intermédiaire agglomérées, principalement de la GFAP. Elles se forment dans les processus astrocytaires (les bras des astrocytes), et apparaissent comme des dépôts dans la cellule. Ces agrégats sont parfois retrouvés dans d’autres types de maladies, mais pas avec l’abondance, ou la distribution particulière dans le cerveau, qui sont observées dans la maladie d’Alexander. Les inclusions de fibres de Rosenthal sont visibles sur des colorations du cerveau. Elles ressemblent à des faisceaux épais, allongés, semblables à des vers ou en forme de tire-bouchon, mesurant environ 10 à 40 micromètres de diamètre sur 100 micromètres de longueur.
Aujourd’hui, ce qui déclenche la formation des fibres de Rosenthal n’est pas clair, et il n’est pas établi que ces fibres soient protectrices ou toxiques. Des recherches supplémentaires sont nécessaires pour comprendre le rôle spécifique de GFAP et les mécanismes responsables de la maladie.
3. L’expression clinique
La maladie d’Alexander a été identifiée en 1949 par le docteur Stewart Alexander, pathologiste australien, qui le premier décrivit un cas infantile présentant des fibres de Rosenthal et une perte de myéline. C’est une leucodystrophie neurodégénérative, ce qui signifie que, en parallèle de la perte de substance blanche, les neurones perdent continuellement leur structure et leur fonctionnalité au fil du temps.
Les symptômes de la maladie d’Alexander peuvent varier. Ils dépendent largement de l’âge d’apparition de la maladie mais peuvent inclure : des spasmes, des troubles d’apprentissage, des problèmes d’alimentation, une taille de tête et de cerveau accrue, une hydrocéphalie (présence de liquide dans le cerveau), un retard de développement et de croissance, des crises épileptiques, une mobilité réduite, des problèmes d’élocution, de régression mentale, des difficultés à avaler, une incapacité à tousser, des troubles du sommeil.
La maladie d’Alexander chez le petit enfant
Dans 80% des cas, la maladie d’Alexander se manifeste avant l’âge de 3 ans. Les petits patients développent une hypertrophie du cerveau et de la tête (c’est-à-dire une augmentation de volume), des convulsions, une raideur des bras et / ou des jambes, une déficience intellectuelle et un retard de développement.
Dans de rares cas, une forme néonatale de maladie d’Alexander survient au cours du premier mois de vie et est associée à une déficience intellectuelle sévère et à un retard de développement, à une accumulation de liquide céphalo-rachidien dans le cerveau (hydrocéphalie) et à des convulsions.
La maladie d’Alexander chez l’enfant plus âgé
Quatorze pour cent des patients développent les premiers symptômes entre 3 et 12 ans. Pour ces formes juvéniles les symptômes comprennent généralement une mauvaise coordination (ataxie), des difficultés à avaler, des problèmes d’élocution, des crises d’épilepsie
La forme adulte de la maladie d’Alexander
Chez les patients atteints par la forme adulte de la maladie d’Alexander (6% des cas), les symptômes neurologiques et le pronostic sont variables. Les symptômes ne sont pas spécifiques et comprennent, comme pour les formes juvéniles, une mauvaise coordination (ataxie), des difficultés de déglutition, des anomalies de la parole, et des convulsions.
En règle générale, la maladie d’Alexander est moins grave lorsqu’elle se développe à l’âge adulte. La taille de la tête et la capacité mentale peuvent être complètement normales à ce stade, mais un lent déclin mental est possible.
Maladie d’Alexander chez les personnes âgées (65 ans et plus)
Il est extrêmement rare que la maladie d’Alexander se développe aussi tardivement. Si tel est le cas, les symptômes sont souvent confondus avec ceux d’une sclérose en plaques ou ceux d’une tumeur au cerveau. La gravité de la maladie est alors souvent si modérée, que la maladie d’Alexander est diagnostiquée après le décès, lorsqu’une autopsie révèle des dépôts de protéines inhabituels dans le cerveau.
Classification symptomatique
Des systèmes de classification plus récents ont été proposés qui reposent davantage sur l’emplacement des lésions dans le cerveau et la moelle épinière et, par conséquent, sur les types de symptômes.
Les patients de type I ont une prédominance frontale des lésions, tous sont précoces et ont une évolution plus agressive de la maladie. En règle générale, les patients de type I présentent divers retards de développement, affectant à la fois les capacités cognitives et motrices (telles que le langage ou la marche), suivis d’une perte de jalons, d’une augmentation anormale de la taille de la tête et souvent de convulsions.
Les patients de type II ont une prédominance du cerveau postérieur, avec des apparitions tout au long de la vie, et une progression plus lente de la maladie. Les patients de type II éprouvent plus de problèmes de marche. De nombreux patients ont des problèmes de vomissements excessifs, de difficulté à avaler et à parler
Les patients de type I prédominent dans la littérature, mais cela reflète probablement un biais de vérification, car les patients adultes en particulier sont souvent mal diagnostiqués avec d’autres conditions, telles que la maladie de Parkinson ou la sclérose en plaques.
4. Le diagnostic de la maladie
Bien qu’il existe des symptômes et des éléments cliniques pouvant orienter vers une maladie d’Alexander, le diagnostic ne peut être validé que par des tests génétiques. Les personnes présentant des symptômes sont référées à un spécialiste de la génétique et du métabolisme qui peut s’assurer qu’un diagnostic correct est posé.
5. Le conseil génétique
A ce jour, plus de 100 mutations génétiques différentes ont été identifiées. Dans la plupart des cas, la mutation génétique à l’origine de ce trouble est spontanée, ce qui signifie qu’il ne s’agit pas d’une mutation transmise par l’un ou l’autre des parents. Par conséquent, il est peu probable que d’autres enfants des mêmes parents aient la maladie d’Alexander. Il est peu probable que les frères et les sœurs d’enfants atteints de la maladie d’Alexander soient eux aussi atteints de la maladie. Seuls quelques cas familiaux ont été rapportés. Il est important de parler avec un généticien ou un expert de la maladie d’Alexander pour discuter de la probabilité que la maladie soit transmise génétiquement à d’autres membres de la famille.
6. Les traitements
A ce jour, il n’existe pas de traitement définitif de la maladie d’Alexander. Cependant, quelle que soit la forme diagnostiquée, il est important de mettre rapidement en place des soins médicaux complets. Grâce à des soins médicaux proactifs et complets, les patients peuvent éviter des souffrances et des complications inutiles, et avoir la meilleure qualité de vie possible.
Une attention particulière doit être portée :
• Aux soins généraux
• Ergothérapie et physiothérapie
• Nutrition
• Orthophonie
• Antibiotiques pour toute infection développée
• Médicaments antiépileptiques pour contrôler les crises
La recherche
Avec l’absence d’un traitement définitif, la recherche continue. Elle est doit encore mieux comprendre la maladie afin d’envisager des traitements.
• Oligonucléotides antisens
Pour traiter la maladie d’Alexander, les chercheurs réfléchissent à l’utilisation d’oligonucléotides antisens (ASO), une technologie permettant de supprimer l’expression d’un gène en particulier. Ces traitements sont en cours de développement dans plusieurs maladies, et un premier ASO a été autorisé en Europe et aux Etats-Unis pour le traitement de l’amyotrophie spinale, une maladie rare neuromusculaire.
Pour la maladie d’Alexander, un traitement par ASO est déjà à l’étude chez l’animal, et les premiers résultats sont encourageants. Une seule injection d’ASO dans le ventricule cérébral agit en quelques semaines. Les fibres de Rosenthal disparaissent et plusieurs marqueurs reviennent à des niveaux proches de la normale. Ces résultats ont suscité un intérêt important dans la communauté clinique et pourraient conduire à un essai clinique formel mais il reste encore beaucoup à faire, notamment évaluer la capacité des ASO à produire des améliorations des phénotypes moteurs et comportementaux.
• EJP Alexander
Dans le cadre de sa mission de soutien à la recherche contre les leucodystrophies, ELA international participe à un Programme Conjoint Européen pour les maladies rares (EJP Maladies Rares) portant sur la maladie d’Alexander. L’objectif de ce projet de 3 ans, est de mieux comprendre le processus de déclenchement et de développement de la maladie. Il est conduit par le professeur Elly Hol de l’université d’Utrecht aux Pays Bas et associe 7 équipes de recherche de 6 pays différents (Suède, République Tchèque, Israël, Espagne, Luxembourg, Nouvelle Zélande). La synergie des expertises et des connaissances, et la valorisation des ressources au-delà des frontières font la force de ces programmes européens auxquels ELA s’associe.
Conclusions
De nombreuses zones d’ombre existent encore autour de la maladie d’Alexander tant du point de vue de la compréhension des mécanismes de la maladie, que de la recherche de traitements. Seuls 500 cas ont été décrits dans le monde ce qui en fait une maladie extrêmement rare et complique la recherche. Aujourd’hui les médecins s’attachent à mieux prendre en charge ces patients par des soins médicaux proactifs et globaux, et à offrir la meilleure qualité de vie possible pour ces patients. La recherche continue, avec le soutien d’ELA, pour espérer demain des solutions.
[1] Cytosquelette : ensemble organisé des polymères biologiques d’une cellule qui lui confèrent l’essentiel de ses propriétés mécaniques.